This site outlines the clinical trial research governance requirements for UNSW staff, students and researchers. Feedback on the clinical trials research governance process can be submitted by email to clinicaltrials@unsw.edu.au or directly to the Director RECS, Dr Ted Rohr E: ted.rohr@unsw.edu.au.

At UNSW Clinical Trials are defined as any research study that prospectively assign human participants or groups of humans to one or more health-related interventions (which may include placebo or other control) where the primary aim of the research is to evaluate the effect of the intervention on health outcomes.
The Clinical Trial Decision Tool can be used to assist researchers in determining whether research meets the UNSW definition of a clinical trial.
The sponsor of a trial is the company, institution or organisation which takes responsibility for the initiation, management, and/or financing of a clinical trial. The main types of clinical trial sponsors are:
- Commercial Sponsor
- Contract Research Organisation Sponsor
- Collaborative Research Group Investigator Initiated Sponsor
Clinical Trials where UNSW is responsible as sponsor
UNSWs role as clinical trial sponsor is not automatically confirmed. UNSW researchers and staff members must obtain written confirmation from the UNSW Sponsors Delegate before recruitment and data collection for a clinical trial commences. UNSW can assume the role of trial sponsor for clinical trials conducted within Australia that meet UNSWs criteria for trial sponsorship. A list of the criteria is specified on the Sponsor Related Responsibilities webpage.
It is recommended that the following is completed before a clinical trial is submitted to a Human Research Ethics Committee for ethical approval to avoid further amendment or modification requests:
- A UNSW Clinical Trial Protocol Templates is used to develop the trial protocol.
The draft Clinical Trial Protocol is submitted for review by the UNSW Sponsors Delegate before submitting it for review and approval by a Human Research Ethics Committee.
Utilising an unapproved therapeutic good or an approved therapeutic good outside of the approved indications or unapproved purpose requires a Clinical Trail Notification through the Therapeutic Goods Administration (TGA) portal. The UNSW Clinical Trials Governance office manages the Therapeutic Goods Administration (TGA) Clinical Trial Notification (CTN) submission portal and the notification and submission requirements as outlined on the UNSW Submitting a Clinical Trial Notification to the TGA page.
The sponsor of a trial must enter into a clinical trial research agreement (CTRA) with each site, documenting the obligations of each party with respect to the conduct of the trial and outlining any payments that will be made to the site.
Information on how to develop the clinical trial research agreement request requirements, clinical research agreement information, submission instructions and the time involved in the process can be found on the Clinical Trial Research Agreement Request for Signature page.
Insurance policies for clinical trials are not automatically covered. Therefore, researchers are required to confirm cover with UNSW Insurance. In addition, the following documentation must be prepared and sent via email to financehelp@unsw.edu.au with clinicaltrials@unsw.edu.au copied on the submission email:
- Clinical Trials Insurance Application.
- Clinical Trial Protocol (a draft protocol is acceptable if the trial is pending approval by an HREC)
UNSW Insurance will confirm insurance or seek further clarifications via return email. A copy of this confirmation (if clinicaltrials@unsw.edu.au) is not copied on the return email must be sent to clinicaltrials@unsw.edu.au.
Confirmation of clinical trials insurance cover must be provided before the following:
- Recruitment or Data Collection Commences.
- Clinical Trial Research Agreements are signed.
Insurance cover confirmation does not need to be provided on multiple occasions or each time a site clinical trial research agreement is submitted for signing.
A clinical trial protocol provides the plan for the undertaking of a clinical trial. The ICH GCP-guided requirements for the development of a clinical trial protocol are outlined at this section. The following templates are provided for use:
- Clinical Trial Protocol Template [ Medical Device]
- Clinical Trial Protocol Template [Medical Product]
- Clinical Trial Protocol Template [Health Interventions]
If UNSW agrees to be the sponsor for an international clinical trial, it is a requirement that an overseas clinical trial protocol template must be used. It is recommended that the standard sections such as delegation of sponsor duties, safety monitoring reporting, protocol deviations, serious breach, data ownership and risk based monitoring are included or added as an addendum to the protocol.
Clinical trials that require a clinical trial protocol
A clinical trial protocol that complies with the Good Clinical Practice Guideline is to be developed for:
- Clinical trials that involve an investigational therapeutic medical product, device or biological;
- Clinical trials that require notification via the Therapeutic Goods Administration or the relevant regulatory body within the country that the trial will be conducted in; and/or
- Clinical trials where the trial sponsor requires a protocol.
A protocol is not required but is recommended for:
- Clinical trials involving a medical product, device or biological being used for the intended purpose listed in their respective Australian Register of Therapeutic Goods Public Summary or as per its conditions of approval in the country that the trial is being conducted;
- Clinical trials that do not involve a medical product, device or biological; and/or
- Trials that do not require notification via the Therapeutic Goods Administration Clinical Trial Notification process or the relevant regulatory body within the country that the trial will be conducted in.
Clinical Trial Protocol requirements for trials where UNSW is to be the Sponsor
The contents of the clinical trial protocol must include all items included at section 6.1 of the Guideline for Good Clinical Practice E6(R2). In particular, the following items must be included as distinct sections in the clinical trial protocol:
Risks
- The risks relating to the investigational medical product, device, biological, trial conduct, design and methods must be defined in the clinical trial protocol.
Protocol Deviations and Serious Breaches
- Protocol deviation and serious breach definitions, procedures for recording, reporting and developing corrective and preventative action plans must be documented in the clinical trial protocol and reflect the requirements outlined in the protocol deviations and serious breach section.
Safety Monitoring
- Safety monitoring definitions, procedures for recording, reporting, and assessing safety monitoring reports must be documented in the clinical trial protocol and reflect the requirements outlined in the safety monitoring section.
Delegation of duties
The UNSW Clinical Trial Delegation Log defines the trial responsibilities of the UNSW Sponsor’s Delegate, the Coordinating Principal Investigator(s), Site Principal Investigator(s) and any trial-related personnel that accompany the clinical trial protocol.
The delegation's log must specify that the responsibilities for the conduct, oversight and monitoring for the trial are delegated to the Coordinating Principal Investigator.
The Coordinating Principal Investigator may delegate trial-related responsibilities to the approved Principal Investigator(s) or trial related personnel. All trial-related duties delegated by the Coordinating Principal Investigator or Principal Investigator(s) and/or trial related personnel are only to be delegated to those that are qualified by experience and training.
The delegations log must specify that the UNSW Sponsor’s Delegate is notified of:
- Protocol Deviation reports outlined in the UNSW Research Misconduct Procedure.
- Any serious breach of Good Clinical Practice, the clinical trial protocol, the clinical trial standard operating procedures, or the human ethics approval that is likely to affect to a significant degree the safety or rights of participants or the reliability and robustness of the data generated in the clinical trial.
- Suspected Unexpected Adverse Reactions (SUSAR) or Unanticipated Serious Adverse Device Effect (USADE) events
- Significant Safety Issues likely to (or have the potential to) affect to a significant degree the safety or rights of participants or the reliability and robustness of the data generated in the clinical trial
- Urgent Safety Measures implemented to remove or prevent a significant safety issue
- Safety Reports relating to the continuation, suspension, or discontinuation of the clinical trial for safety reasons as they arise.
- Participant complaints or concerns received in relation to the conduct of the research.
- Any significant modifications to the clinical trial that are likely to affect to a significant degree the safety or rights of participants or the reliability and robustness of the data generated in the clinical trial.
- Amendments to clinical trial research agreements or service level agreements.
- Revisions to regulatory requirements including correspondence with the Therapeutic Goods Administration, clinical trial registries or the FDA.
- Site authorisation, addition of new sites, closure of approved sites of changes to the site Principal Investigator that is responsible for a site.
Monitoring of compliance
- Procedures for monitoring compliance must be documented in the trial protocol.
A trial should be conducted in compliance with the clinical trial protocol that has received prior Human Research Ethics Committee and, where applicable, overseas Institutional Review Board approval or favourable opinion.
A Human Research Ethics Committee/Institutional Review Board must be selected to complete an ethical review of the clinical trial. If the research requires the recruitment of participants from private institutions, public hospitals or sites in other countries, multiple HRECs or IRBs may need to be identified to review and approve a clinical trial. Trials conducted in Australia where UNSW is the sponsor must be reviewed and approved by an Australian Human Research Ethics Committee formally registered with NHMRC.
Specific details identifying the reviewing/approving HRECs/IRBs and the sites covered by the ethics approval must be included in the clinical trial protocol.
Approvals established with an NHMRC-recognised HREC outside of UNSW must be submitted for noting via the UNSW external ethics approval process. Clinical trials conducted overseas require UNSW HREC approval before a submission is made to the in-country IRB or equivalent.
ICH Good Clinical Practice (GCP) is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human subjects. Compliance with this standard provides public assurance that the rights, safety and well-being of trial subjects are protected, consistent with the principles that have their origin in the Declaration of Helsinki, and that the clinical trial data are credible.
Clinical Trials Involving Investigational Therapeutic Goods
The therapeutic goods legislation requires that the use of investigational therapeutic medical products or devices in a clinical trial conducted under the CTN/CTX schemes are designed, conducted and monitored in accordance with the Guidelines for Good Clinical Practice (E6, R2), the Australian National Statement on Ethical Conduct in Human Research, and the clinical trial protocol as approved by the reviewing Human Research Ethics Committee (or Institutional Review Board as known in the US and other countries).
It is the responsibility of the Coordinating and Principal Investigators to ensure that all investigators and trial-related staff have current ICH Good Clinical Practice Training. Certificates of training completion need to be stored as an GCP essential document. According to ICH GCP, clinical trial staff are required to update their GCP training at least once every 3 years.
It is the responsibility of the Coordinating and Principal Investigators to familiarise themselves with the requirements of the Guideline for Good Clinical Practice (E6, R2).
Other Clinical Trials
It is recommended that the requirements of GCP are met for all other trials involving human participants outside the strict definition of a clinical trial per se. The recommendation refers to researchers' recognition that best practice leads to good research outcomes.
Documenting GCP Training
Records of Good Clinical Practice Certificates from an accredited training authority must be documented for all investigators and personnel conducting clinical trial activities. Therefore, it is the responsibility of the Coordinating Principal Investigator to retain records of Good Clinical Practice certification and make them available to the UNSW Sponsors Delegate if requested.
The Investigator's Brochure (IB) is a compilation of the clinical and nonclinical data of the investigational product(s) that are relevant to the study of the product(s) in human subjects. Its purpose is to provide investigators and others involved in the trial with the information to facilitate their understanding of the rationale for, and their compliance with key features of the protocol, such as the dose, dose frequency/interval, methods of administration and safety monitoring procedures.
Clinical trials that require an investigator brochure
An investigator brochure that complies with the Good Clinical Practice Guideline must be developed for:
- Clinical trials that involve an investigational medical product, device or biological where this information has not been included in the clinical trial protocol.
- Clinical trials that require notification via the Therapeutic Goods Administration or the relevant regulatory body within the country that the trial will be conducted in.
- Clinical trials where the trial sponsor requires an investigator brochure.
Clinical trials that do not require an investigator brochure
An investigator brochure is not required but can be submitted for:
- Clinical trials involving a medical product, device or biological being used for the intended purpose listed in their respective Australian Register of Therapeutic Goods Public Summary or as per its conditions of approval in the country that the trial is being conducted.
- Clinical trials that do not involve a medical product, device or biological.
- Trials do not require notification via the Therapeutic Goods Administration Clinical Trial Notification process or the relevant regulatory body within the country that the trial will be conducted in.
The sponsor is responsible for developing procedures for identifying, assessing, and selecting the site(s), institution(s) and investigator(s) that are to participate in the clinical trial.
For trials sponsored by UNSW, the Coordinating Principal Investigator will be responsible for selection of trial sites, site principal investigator(s) and for the completion of feasibility activities.
The Coordinating Principal Investigator must ensure that the following is in place for each participating site:
Site Principal Investigator
- The Principal Investigator is qualified by experience and training to provide oversight and conduct the clinical trial at the site.
- The Principal Investigator has expertise with the target population and disease area being studied.
- The Principal Investigator has or is to be provided with protocol specific training and is aware of the requirement to conduct the trial in accordance with the protocol, HREC approved procedures and GCP.
Institution
- The institution responsible for the site has research governance. procedures in place for authorising the commencement of a clinical trial.
- The institution responsible for the site has adequate facilities, resources, technology, and qualified personnel to conduct the trial in accordance with the protocol, HREC approval and where applicable GCP.
- The institution has procedures in place for facilitating the review of clinical trial research agreements, service agreements, insurance and indemnity requirements.
- The institution responsible for the site has procedures in place for responding to participant complaints, allegations of research misconduct and/or serious breaches of GCP.
Site
- The site has access to the target population to meet recruitment targets.
Site Personnel
- All site personnel responsible for conducting the clinical trial in accordance with the protocol are qualified by experience and training.
- Copies of CVs, Good Clinical Practice training certificates and records of qualifications to demonstrate experience by qualification and training are obtained and documented.
The Coordinating Principal Investigator must register the clinical trial on a publicly accessible register complying with international standards before the recruitment of the first participant. Below are the recommended platforms to register clinical trials.
Information to assist with classifying the risk profile of clinical trials and the monitoring requirements can be accessed on the following webpage:
This section outlines the requirements and definitions for safety reporting.
Safety Monitoring Reporting
Adverse Event (AE), Adverse Device Effect (ADE), Adverse Reaction (AR), Serious Adverse Event (SAE)/Serious Adverse Reaction (SAR), Serious Adverse Device Effect (SADE), Suspected Unexpected Serious Adverse Reaction (SUSAR), Unanticipated Serious Adverse Device Effect (USADE), Significant Safety Issue (SSI) and Urgent Safety Measure (USM) are to be reported by the principal site investigator to the Sponsor. If UNSW is the Sponsor, the site principal investigator report must be sent to the Coordinating Principal Investigator.
Safety Monitoring Report Form
A form used to report events must be designed for the trial. Paper-based versions or electronic versions can be developed. The forms should include a mechanism for assessment of seriousness, causality, and expectedness to be made.
Assessment and Management of Safety Monitoring Reports
Once a report has been made, procedures for assessing and classifying reports following the safety reporting assessment flowchart must be developed and documented in the protocol before recruitment and data collection starts. If UNSW is the Sponsor, the Coordinating Principal Investigator must develop the following procedures to meet these requirements.
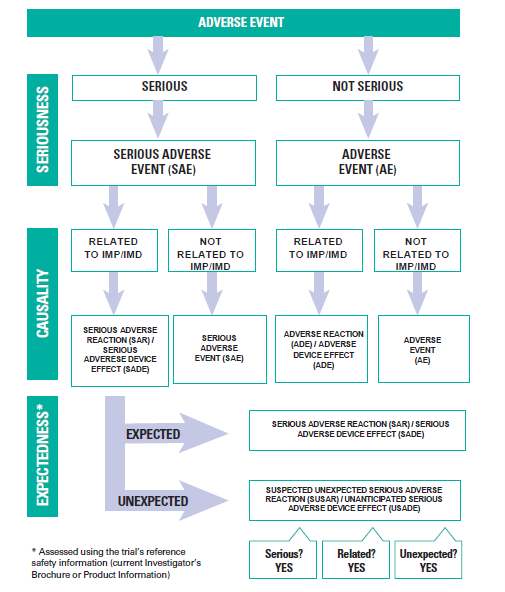
Adverse Event Report Assessments
Event report forms classified the safety monitoring event as "not serious" must be assessed by a Qualified Physician named in the clinical trial protocol. The Qualified Physician must use the Safety Reporting Assessment Flowchart to determine whether the event is an adverse reaction or an adverse event. The Qualified Physician cannot delegate this responsibility to other research personnel. Please note that non IMD/IMP Clinical Trials do not require a Qualified Medical Physician to provide oversight over the trial however adverse events should be reviewed by Trial Monitoring Group that should consist of a physician. The reporting timeframes for adverse event reports from participating sites to the Coordinating Principal Investigator must be specified in the protocol. All adverse event reports must be recorded in the safety monitoring register.
Definition of a Qualified Physician/Medical Expert: A medical expert or clinician that is a listed investigator who has experienced by qualification and training in the research area that provides safety oversight, such as the ongoing monitoring of reports of adverse events (AEs) submitted by investigational sites to identify safety concerns. The Qualified Physician/Medical Expert cannot be a Principal Investigator responsible for a trial site. The name of this person must be specified in the general information section of the protocol. The role cannot be delegated to another party.
Serious Event Report Assessments
The Sponsor's Medical Expert must assess event report forms that classify the event as "serious". Therefore, a report must be sent to the Sponsor's Medical Expert for assessment as soon as possible and within seven working days of receiving the report form. The following criteria classify the report as serious:
- Fatal
- Life-threatening illness or injury (actual risk of death at the time of the event)
- Permanent impairment of body structure or body function
- In-patient or prolonged hospitalisation (not for a pre-existing condition or an elective surgery)
- Medical or surgical intervention to prevent life-threatening illness or injury or permanent impairment to a body structure or function
- Led to fetal distress, fetal death or congenital abnormality or birth defect.
The Sponsor's Independent Medical Expert must use the Safety Reporting Assessment Flowchart to determine whether the event is a serious adverse reaction, a serious adverse event, or a serious adverse device effect.
The Sponsor's Independent Medical Expert cannot delegate this responsibility to other research personnel. Therefore, all decisions made by the Sponsor's Medical Expert are to be recorded.
Sponsor's Independent Medical Expert
A person who is independent of the listed investigators that is a qualified medical physician or clinician who is qualified by experience and training in the research area that provides safety oversight, such as the ongoing monitoring of reports of serious adverse events (SAEs) submitted by investigational sites to identify safety concerns and make recommendations for continuing or stopping a trial.
Serious Adverse Reaction (SAR) or Serious Adverse Device Effect (SADE)
Any serious adverse events related to the investigational medical product or device that occur and are referenced in the clinical trial protocol, the product information, referenced safety information or the investigator brochure must be classified by the Sponsor’s Independent Medical Expert as a SAR or a SADE.
Suspected Unexpected Adverse Reaction (SUSAR) or Unanticipated Serious Adverse Device Effect (USADE)
Any serious adverse events related to the investigational medical product or device that occur and are not referenced in the clinical trial protocol, the product information or investigator brochure must be classified by the Sponsors Independent Medical Expert as a SUSAR or USADE.
Register of Clinical Trial Safety Monitoring Reports
A register of all event reports made and assessed is to be kept by the Sponsor. If UNSW is the Sponsor, the Coordinating Principal Investigator (or delegated staff member as reflected in the trial delegation log) must maintain a register of safety monitoring events.
Reporting of Clinical Trial Safety Monitoring Reports
For trials where UNSW is the Sponsor, single case reports of Adverse Events (AEs), Adverse Reactions (AR), Adverse Device Effect, Serious Adverse Events (SAEs) Serious Adverse Reactions (SARs), Serious Adverse Device Effects (SADEs) reports do not need to be reported to the UNSW Sponsor's Delegate. However, all single case reports must be recorded in a safety monitoring register and need to be reported to the UNSW Sponsor's Delegate annually. The UNSW Safety Monitoring Register Template UNSW is provided as an example of the information to be recorded in a register.
The Trial Management Group (TMG), Trial Safety Committee (TSC) or the Data Safety Monitoring Board (DSMB) must review the safety information to identify any serious emerging safety concerns. If safety concerns are identified, a plan to minimise the time participants may be placed at excess risk of harm must be developed. If no emerging safety issues are identified, the TMG must provide a safety review summary at least annually to the Clinical Trial Governance Office. In addition, the following must be reported to the UNSW Sponsor's Delegate via email to clinicaltrials@unsw.edu.au:
- A brief analysis of the safety profile of the trial intervention(s) and its implications for participants considering all available safety data and the results of relevant clinical or non-clinical studies (annually).
- A brief discussion of the implications of the safety data to the trial's risk-benefit ratio
- A description of any measures taken or proposed to minimise risks. (annually).
- Copies all safety review summaries along with the line listings that relate to these summaries.
- A report of emerging safety issues or where no safety issues have been identified documented evidence (e.g., minutes of a meeting) confirming that a review was undertaken.
- Plan to minimise the time participants are placed at excess risk due to emerging safety issues (within seven days of becoming aware of the safety issues).
Reporting of Suspected Unexpected Adverse Reactions (SUSAR)
Any clinical trial safety monitoring reports classified by the Sponsor's Medical Expert as a SUSAR must be reported to the Coordinating Principal Investigator.
The Coordinating Principal Investigator must report to the TGA and the UNSW Sponsor's Delegate:
- Any fatal or life-threatening Australian SUSARs or USADEs immediately, but no later than seven calendar days after being made aware of the case, with any follow up information within a further eight calendar days.
- All other Australian SUSARs or USADEs, no later than 15 calendar days after being made aware of the case.
Significant Safety Issues (SSIs)
Significant safety issues are defined as any safety issue that could adversely affect participants' safety or materially impact the continued ethical acceptability or conduct of the trial. In addition, the following steps must be followed in situations where urgent measures are taken to eliminate an immediate hazard to a participant's health or safety.
Principal Investigators must report significant safety issues must be reported immediately to the UNSW Sponsor’s Delegate. The report must be supported by advice from the principal Investigator addressing the following points:
- A description of the significant safety issue and any urgent safety measure implemented.
- A description of measures taken and advice on whether a temporary halt of the trial is required for safety reasons.
- An amendment of the clinical trial protocol needs to be submitted to the approving HREC.
Reporting of Significant Safety Issues (SSIs) and Urgent Safety Measures
The Principal Investigator is to report any significant safety issues/urgent safety measures to the Institution within 72 hours of becoming aware of the event. The UNSW Sponsor’s Delegate will then ensure that the significant safety issue is reported to the TGA, HREC and investigators within 15 calendar days of the sponsor instigating or being aware of the issue. For Urgent Safety Measures the Sponsor’s Delegate will notify the TGA, HREC and investigators within 72 hours.

This section outlines the safety monitoring requirements for clinical trials that do not involve an investigational medical product or device.
Safety Monitoring Reporting
Safety event reports are to be reported by the principal site investigator to the sponsor. If UNSW is the sponsor, the Coordinating Principal Investigator must develop the following procedures to meet these requirements. If the trial does not have any participating sites, a process for recording and documenting adverse events and incidents is to be detailed as a procedure or within the HREC approved application.
Adverse Event: An adverse event is defined as any untoward occurrence (medical or other) in a clinical trial participant administered one or more of the trial interventions. All expected or known discomforts, harms, or effects that will be (or have the potential to be) induced by the trial procedures must be specified in the Human Ethics Application or a separate safety monitoring procedure. The safety information must also include:
- Measures for minimising the risk of a participant experiencing an event
- A plan for providing support of care for participants that experience an event.
Serious adverse event: A serious adverse event is any event that results in death, is life-threatening, requires hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability or incapacity, or is a congenital anomaly or birth defect. The clinical trial protocol must outline the research team's process to assess whether the event was related to the clinical trial, its procedures, or the interventions.
Significant Safety Issue: Defined as an issue that could adversely affect participants' safety or materially impact the continued ethical acceptability or conduct of the trial. The human ethics application of safety monitoring procedure must specify that significant safety issues that occur are reported to the UNSW Sponsors Delegate immediately but no later than seven days.
Urgent Safety Measure: Defined as a measure required to be taken to eliminate an immediate hazard to a participant's health or safety. The human ethics application of safety monitoring procedure must specify that urgent safety measures that occur are reported to the UNSW Sponsors Delegate immediately but no later than seven days.
Safety Monitoring Report Form
For multi-centre trials, sites can use a form to report events that must be designed for the trial. Paper-based versions or electronic versions can be developed. Trials that do not have participating sites can record events in a safety monitoring register.
Register of Clinical Trial Safety Monitoring Reports
A register of all event reports made and assessed is to be kept by the sponsor. For example, if UNSW is the sponsor, the Coordinating Principal Investigator must maintain a register of safety monitoring events. The UNSW Safety Monitoring Register Template UNSW is provided as an example of the information to be recorded in a register.
Assessment of Safety Monitoring Reports
A group or committee must be established to assess safety monitoring reports. The group or committee must include at least one person qualified by experience and training who can provide an independent assessment of serious adverse events, significant safety issues, and urgent safety measures.
Adverse event reports can be assessed by an investigator listed in the ethics application that is experienced by qualification and training. The investigator must be independent of the site where the event occurred.
Reporting of Safety Monitoring Reports
Adverse Events and Serious Adverse Events: For trials where UNSW is the sponsor, single case reports of Adverse Events and Serious Adverse Events do not need to be reported to the UNSW Sponsor's Delegate or the HREC. All case reports must be recorded in a safety monitoring register supported by an assessment of trial safety must be reported to the UNSW Sponsor's Delegate annually.
Significant Safety Issues: Are to be reported immediately, but no later than seven days via email to humanethics@unsw.edu.au.
Urgent Safety Measures: Are to be reported immediately, but no later than seven days via email to humanethics@unsw.edu.au.
We are working on procedures to provide meaningful intersections between possible non-compliance with Good Clinical Practice, the protocols approved in the relevant HREC approvals, and definitions of breaches as defined in the Australian Code for the Responsible Conduct of Research.
Protocol Deviation
A protocol deviation is defined as any breach, divergence or departure from the requirements of Good Clinical Practice, the clinical trial protocol, the clinical trial standard operating procedures, or the human ethics approval and does not have a significant impact on the continued safety or rights of participants or the reliability and robustness of the data generated in the research or clinical trial. Protocol deviations are events that do not occur in a persistent or systematic manner, and do not have the potential to result in participant harms. Examples of protocol deviations include but are not limited to:
- Deviations because of participant adherence to the protocol, including rescheduled study visits, participants refusal to complete scheduled research activities or failure to complete self-report questionnaires required by the study protocol.
- Blood samples obtained or clinical trial testing occurring at times close to, but not precisely at the time points specified in the protocol.
- The completion of consent forms, safety monitoring report, case report forms or data collection tools in a manner that is not consistent with the protocol instructions, or failure to make reports within the required reporting timeframes.
- Administration of the clinical trial investigational medical product, or device in a manner that is not consistent with the manufacturer’s instructions for use.
- Use of an unapproved version of the participant information statement or recruitment of participants using unapproved recruitment procedures.
- Inclusion of a participant that does not meet the inclusion criteria.
- An urgent safety measure implemented required to be taken to eliminate an immediate hazard to a participant’s health or safety.
Serious Breach
A serious breach is defined as a breach of Good Clinical Practice, the clinical trial protocol, the clinical trial standard operating procedures, or the human ethics approval that is likely to affect to a significant degree the safety or rights of participants or the reliability and robustness of the data generated in the clinical trial. Examples of serious breaches include but are not limited to:
- Persistent or systematic non-compliance with the instructions for completing consent forms, safety monitoring forms, case report forms or data collection tools that results in continued missed or incomplete data collection.
- Failure to record or report adverse events, serious adverse events, suspected unexpected serious adverse reactions, significant safety issues where urgent safety measures were implemented.
- Failure to conduct clinical trial procedures in accordance with the clinical trial delegation log.
- Widespread and uncontrolled use of protocol waivers affecting eligibility criteria, which leads to harm to trial subjects.
- Failure to report investigational medical product or device defects to the clinical trial sponsor or any relevant regulatory body.
- Failure to conduct research in conformity with the issued approvals, permits or licences in accordance with required laws, regulations, disciplinary standards and UNSW policies relating to the responsible and/or safe conduct of research.
- Concealing or facilitating breaches (or potential breaches) of the Research Code by others.
- Conducting research without the requisite approvals, permits or licences required by laws, regulations, disciplinary standards and UNSW policies related to the responsible and/or safe conduct of research.
- Failure to conduct Research as approved by an ethics review body where that conduct leads to (or has the potential to) results in participant harms.
- Conducting Research without ethics approval as required by the National Statement on Ethical Conduct in Human Research where that conduct leads to (or has the potential to) result in participant harms.
- Any breaches as outlined in the UNSW Research Misconduct Procedure or the Australian Code for responsible conduct of research that leads to (or has the potential to) result in participant harms.
Reporting Protocol Deviations
Protocol deviations occurring at a site must be documented in site files and need to be reported by site principal investigator to the Coordinating Principal Investigator.
The Coordinating Principal Investigator must review the protocol deviation, along with the clinical trial protocol to establish the corrective actions and preventative steps to prevent the deviation from reoccurring.
The protocol deviation and corrective action plan must be reported to the UNSW Sponsor’s Delegate by the Coordinating Principal Investigator or Coordinating Research Team using the protocol deviation report form.
Reporting of a Serious Breach
A serious breach occurring at a participating site must be reported by the site Principal Investigator to the Coordinating Principal Investigator within a specified timeframe.
The Coordinating Principal Investigator must review the serious breach, along with the clinical trial protocol to develop a Corrective and Preventive Action (CAPA) that defines the steps to prevent the serious breach from reoccurring.
The serious breach report and the CAPA is to be provided to the approving HREC and the UNSW sponsors delegate for review and approval.
Reporting of Serious Breaches by Third Parties
A Suspected Breach is a report that is judged by the reporter as a possible serious breach but has yet to be formally confirmed as a serious breach by the sponsor.
A Suspected Breach form must be completed when a third party (e.g. individual / institution) wishes to report a suspected breach of Good Clinical Practice or the protocol. This should be reported directly to the reviewing HREC without reporting through the sponsor.
Recording of Protocol Deviation and Serious Breach Reports
A register of protocol deviation and serious breach reports must be recorded, written records and copies of documentation sent to the sponsor in the must be retained by in the Investigator Site File.
Copies of protocol deviation and serious breach reports must be recorded, written records and copies of documentation sent to the sponsor, referrals made to the HREC or to establish whether a breach of the Australian Code for Responsible conduct of research must be retained in the Master Site File.
Review of a Protocol Deviation and a Serious Breach
The UNSW Sponsor’s Delegate will review reports to establish whether the event meets the definition of a protocol deviation or serious breach, to establish whether the proposed CAPA is appropriate and to establish whether there is or will be an ongoing impact on the reliability and robustness of the data generated.
Advice from the approving HREC will be sought by the UNSW Sponsor’s Delegate on the corrective and preventive actions.
Protocol deviation and/or serious breach reports where a UNSW researcher, staff or student is responsible for the protocol deviation or the serious breach will be reviewed as per the UNSW Research Misconduct Procedure to establish whether a breach of the UNSW Research Code of Conduct has occurred.
Protocol deviation and/or serious breach reports where the UNSW Sponsor’s Delegate determines that the site Principal Investigator(s)/ site personnel are responsible for a protocol deviation or the serious breach will be referred onto their responsible institution for review under their own Research Misconduct procedures to establish whether a breach of the Australian Research Code for the Responsible Conduct of Research has occurred.
Site Authorisation from the responsible institution is to be obtained before recruitment and data collection commences at a site.
The Coordinating Principal Investigator is responsible for ensuring that the following documentation is in place before recruitment and data collection at a trial site commences:
- A letter from a person with the delegated authority for the institution responsible for the participating trial site that authorises the commencement of the trial.
- A clinical trial research agreement signed by the trial sponsor and the delegated authority for the site.
- CVs for all site investigators, and certificates of completed GCP training for all investigators and trial site personnel.
- Evidence to confirm that protocol specific training for investigators and site personnel has occurred.
- Evidence to confirm that site principal investigators and trial site staff have been made aware of the requirement to maintain adequate and accurate source documents and trial records that include all pertinent observations on each of the site’s trial subjects. Source data should be attributable, legible, contemporaneous, original, accurate, and complete. Changes to source data should be traceable, should not obscure the original entry, and should be explained if necessary (e.g., via an audit trail)
- Evidence to confirm that the site Principal Investigator is aware of all documents to be retained in the Investigator Site File.
- Evidence to confirm that the current HREC approved documentation has been provided to the site.
- Any conflict of interest disclosures has been completed and documented.